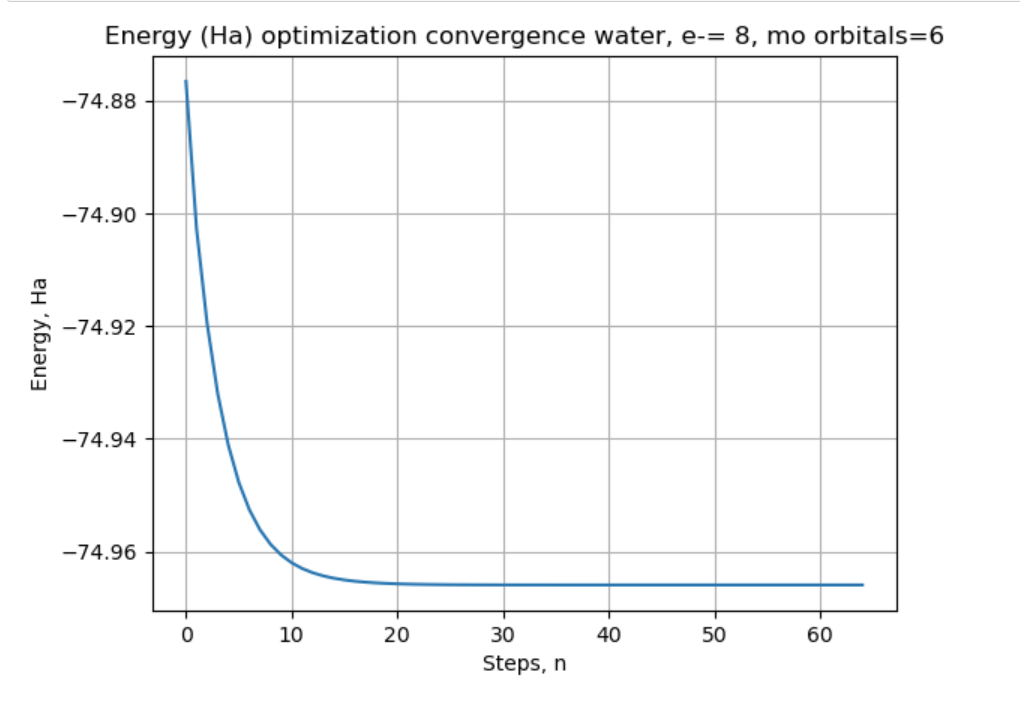
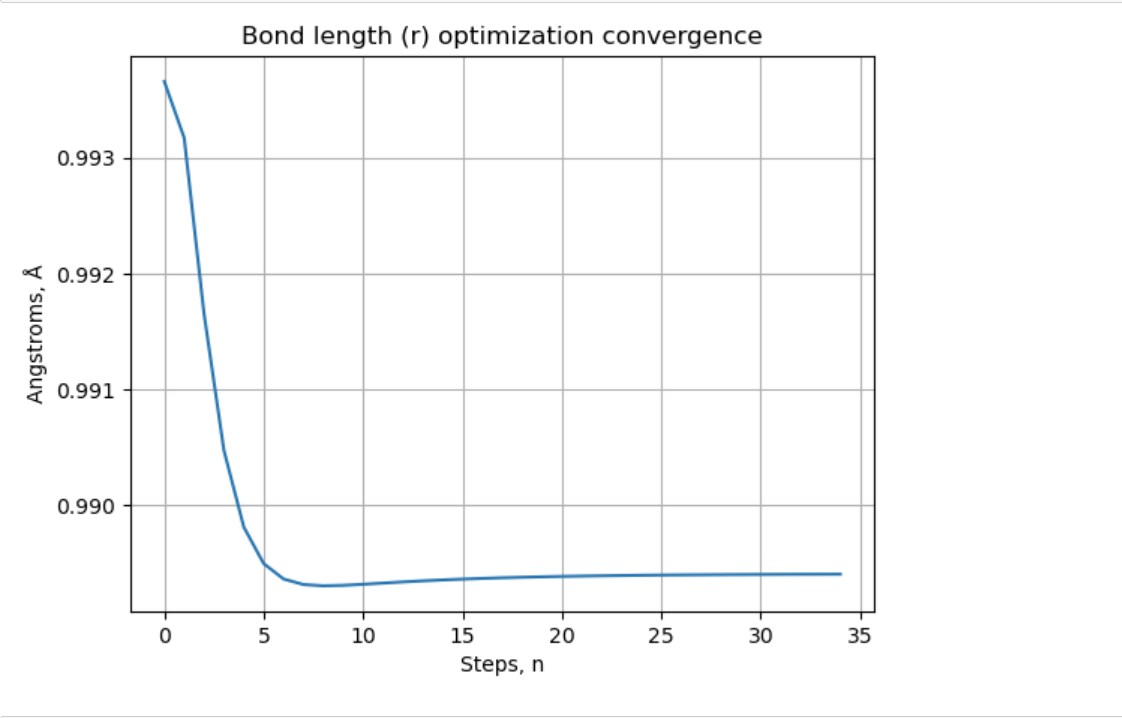
Hello @sjahangiri , given this piece of code:
for n in range(100):
args = [circuit_param, z_matrix]
mol = qml.qchem.Molecule(symbols, geometry)
g_param = grad(energy(mol), argnum = 0)(*args)
circuit_param = circuit_param - 0.25 * g_param[0]
forces = -grad(energy(mol), argnum = 1)(*args)
z_matrix = z_matrix + (0.5 * forces) # forces should be a 2D vector of just the gradient updates of 'r' and 'theta'
geometry = build_geometry(z_matrix[0][0], z_matrix[0][1])
where I’m choosing the optimize water in terms of two parameters ‘r’ and ‘theta’, how would I adapt this code for that? in this piece of code, z_matrix is similar to the geometry vector from the original code, except z_matrix will only store the OH bond length, and the angle betwen both OH bonds, rather then storing all 9 euclidean coordinates of water.
The issue I’m facing is, how would I modify the code so that forces = -grad(energy(mol), argnum = 1)(*args) will return a 2-D vector that is the updates for OH bond length, and OH1-OH2 bond angle, or ‘r’ and ‘theta’ respectively?
Update: I modified the energy(mol) function so that the forces vector is now:
I’m attaching a minimally working example here in case you could kindly take a look at it:
# Minimally working and self-contained example:
# imports and functions:
import pennylane as qml
from pennylane import qchem
from pennylane import numpy as np
from autograd import grad
import time
import math
bohr_angs = 0.529177210903
def unit_vector(vector):
""" Returns the unit vector of the vector. """
return vector / np.linalg.norm(vector)
def angle_between(v1, v2):
""" Returns the angle in radians between vectors 'v1' and 'v2'::
"""
v1_u = unit_vector(v1)
v2_u = unit_vector(v2)
return (np.arccos(np.clip(np.dot(v1_u, v2_u), -1.0, 1.0)) * 180/math.pi) #convert to degrees
def build_geometry(r, angle): # prepares water's xyz coordinates given a bond length and angle
if (not isinstance(r, np.ndarray)):
# If argnum=1 in 'energy(mol)' function, the type of 'r' and 'angle' are for some reason of ArrayBox.
# So to get their values, I use the '._value' attribute but not sure if this is right
# Otherwise, if argnum=0 in 'energy(mol)', then these values are tensor numpy arrays, so no issue
r = r._value
angle = angle._value
bohr_angs = 0.529177210903
r = r/bohr_angs # converts from units of angstroms back to units of Bohrs
x = np.array([[0.10000000, 0.10000000, 0.28377432],
[0.10000000, 1.45278171, -1.00662237],
[0.10000000, -1.45278171, -1.00662237]], requires_grad=True)
# O1's nuclear coordinates, is positioned at x,y,z: (0,0,0)
x[0][0] = 0.0; x[0][1] = 0.0; x[0][2] = 0.0
# H1's nuclear coordinates, is positioned at x,y,z: (r,0,0)
x[1][0] = r; x[1][1] = 0.0; x[1][2] = 0.0
# H2's nuclear coordinates, is position at x,y,z: (r*cos(angle), r*sin(angle), 0)
x[2][0] = r * np.cos(angle * np.pi/180); x[2][1] = r * np.sin(angle * np.pi/180); x[2][2] = 0.0
return x
# Initializations
symbols = ["O", "H", "H"]
geometry = np.array([[0.10000000, 0.10000000, 0.28377432],
[0.10000000, 1.45278171, -1.00662237],
[0.10000000, -1.45278171, -1.00662237]], requires_grad=True)
# Initialize starting r and starting theta
OH1 = geometry[0] - geometry[1]
OH2 = geometry[0] - geometry[2]
r = np.linalg.norm(geometry[0] - geometry[1]) * bohr_angs
theta = angle_between(OH1, OH2)
z_matrix = [np.array([r], requires_grad=True)] + [np.array([theta], requires_grad=True)]
# re-initialize geometry:
geometry = build_geometry(z_matrix[0], z_matrix[1])
mol = qml.qchem.Molecule(symbols, geometry)
dev = qml.device("default.qubit")
def energy(mol):
@qml.qnode(dev, interface="autograd")
def circuit(*args):
# note that active_electrons=2, active_orbitals=2 in this example
qml.BasisState([1, 1, 1, 1, 1, 1, 1, 1, 0, 0, 0, 0], wires=range(12))
# Just apply one gate for simplicity:
qml.DoubleExcitation(*args[0][0], wires=[0, 1, 2, 3])
H = qml.qchem.molecular_hamiltonian(mol.symbols, build_geometry(*args[1][0], *args[1][1]),
active_electrons=8, active_orbitals=6, args=[build_geometry(*args[1][0], *args[1][1])])[0]
return qml.expval(H)
return circuit
circuit_param = [np.array([0.0], requires_grad=True)] * 1
for n in range(1): # Just run 1 iteration
args = [circuit_param, z_matrix] # original line: args = [circuit_param, geometry]
mol = qml.qchem.Molecule(symbols, geometry)
g_param = grad(energy(mol), argnum = 0)(*args)
circuit_param = circuit_param - 0.25 * g_param[0]
forces = -np.array(grad(energy(mol), argnum = 1)(*args)) # without np.array(), this line returns "TypeError: bad operand type for unary -: 'list'""
print("Forces vector:", forces)
z_matrix = z_matrix + (0.5 * forces) # update just 2 parameters
geometry = build_geometry(z_matrix[0][0], z_matrix[1][0]) # re-update geometry from these 2 updated-parameters
print(f'n: {n}, E: {energy(mol)(*args):.8f}, Force-max: {abs(forces).max():.8f}')